
Our research is focused upon these fundamental questions:
· Why and how do neurons die in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)?
· What mechanisms maintain RNA and protein homeostasis in neurons, and how do they fail in disease?
· Can we identify and develop therapies to prevent or block neurodegeneration in both ALS and FTD?
· Why and how do neurons die in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)?
· What mechanisms maintain RNA and protein homeostasis in neurons, and how do they fail in disease?
· Can we identify and develop therapies to prevent or block neurodegeneration in both ALS and FTD?
TDP43 and neurodegeneration in ALS and FTD
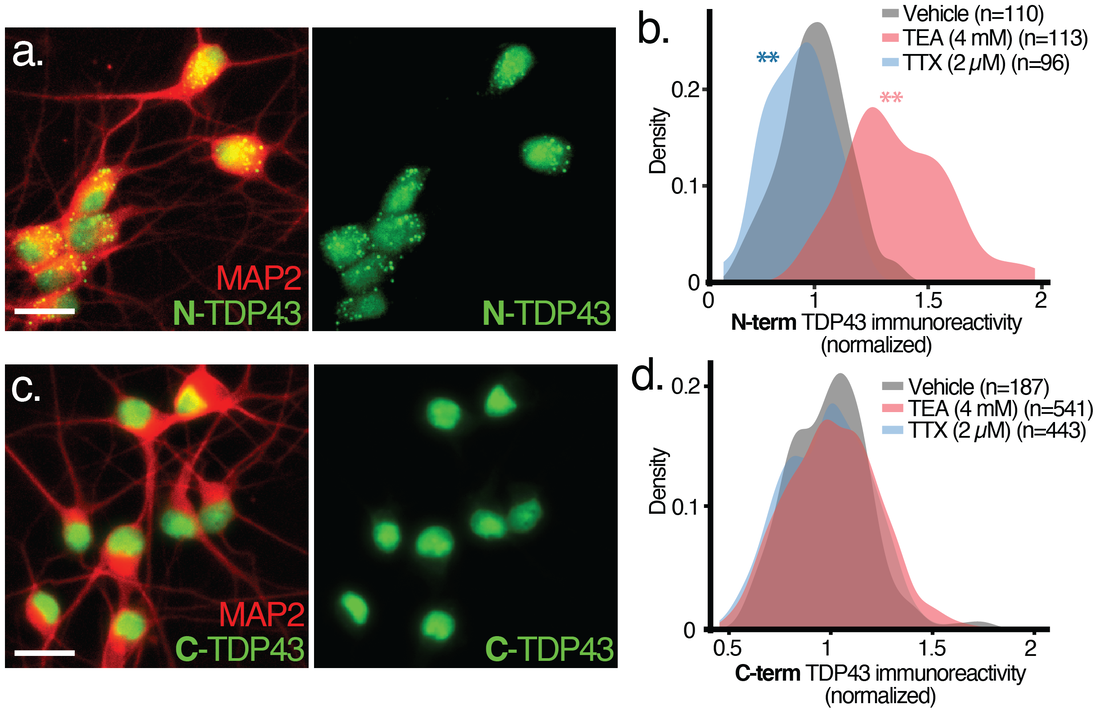
Our work uncovered unusual isoforms of the RNA binding protein TDP43 that are upregulated by neuronal hyperactivity, a conserved feature of ALS. Ongoing experiments seek to define the function, impact and regulation of these isoforms, and whether they represent valid therapeutic targets for ALS and perhaps FTD.
|
Immunostaining using antibodies against N-(a,b) or C-terminal epitopes (c,d) demonstrates bi-directional changes in N- but not C-terminal TDP43 reactivity in human iPSC-derived neurons treated with tetraethylammonium (TEA) to increase activity, or tetrodotoxin (TTX) to block activity.
|

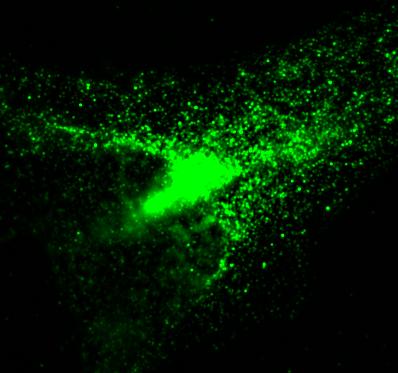
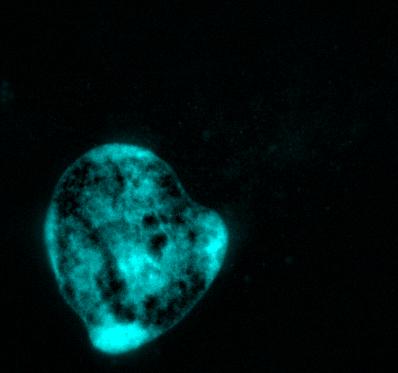
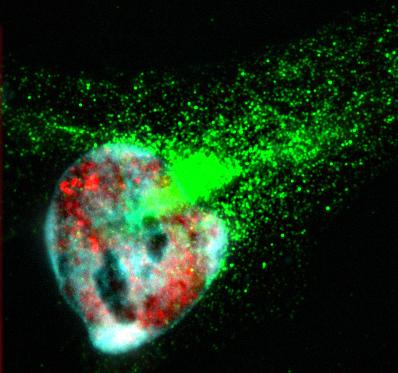
HeLa cells immunostained with antibodies against full-length TDP43 (red) and shortened TDP43 (green) isoforms. Nuclei are stained with Hoechst dye (blue)
Novel approaches to therapy in ALS and FTD
Using automated survival analysis, we survey potential therapeutic strategies through unbiased and targeted approaches.
|
|
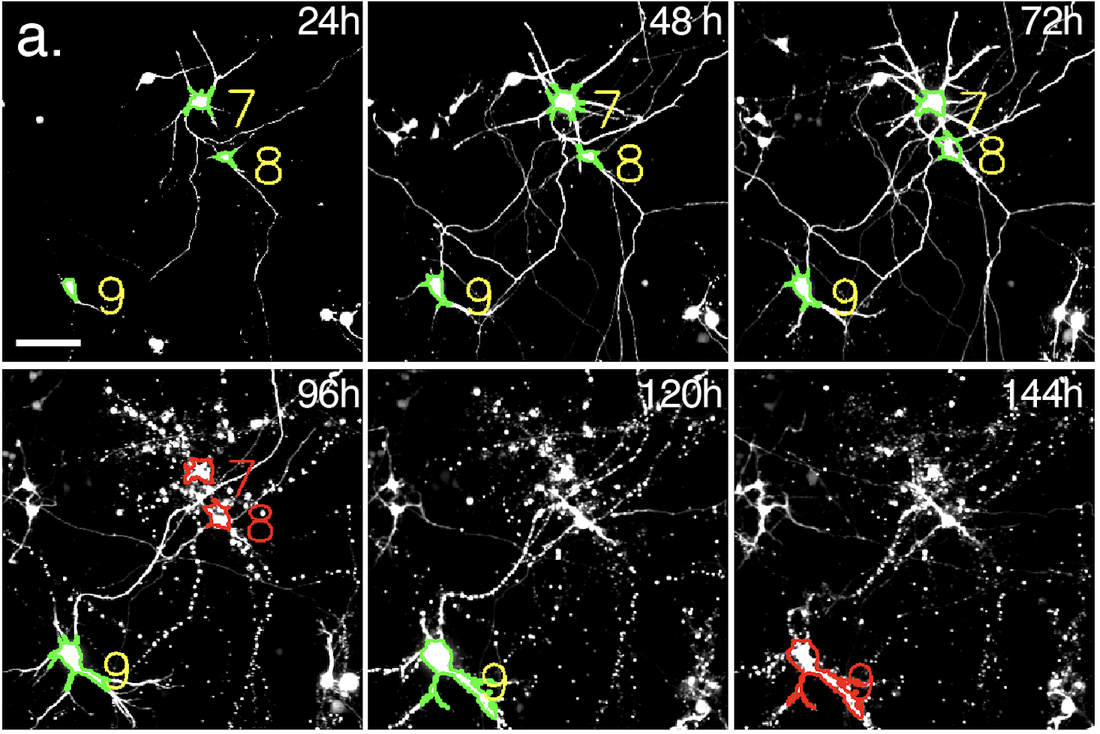
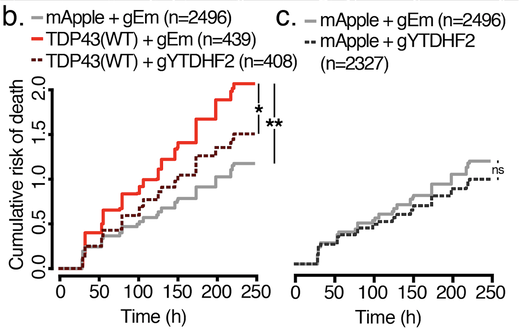
(a) Automated microscopy and survival. Primary rodent cortical neurons expressing fluorescent proteins are tracked over time using custom scripts that label neurons (yellow) and determine their time of death (red). Scale bar, 50µm. (b) Cumulative risk of death plot demonstrating toxicity of low-level TDP43(WT)-mApple vs. mApple alone, and the protective effect of YTHDF2 knockout. gEm, empty Cas9 guide. gYTHDF2, YTHDF2 Cas9 guide. (c) Control neurons were unaffected by YTHDF2 knockout.
RNA decay in ALS and FTD
We observed persistent and pervasive abnormalities in RNA degradation mechanisms in ALS/FTD tissues and in disease models. Ongoing studies seek to determine the origin of these findings and their relevance for disease pathogenesis.
|
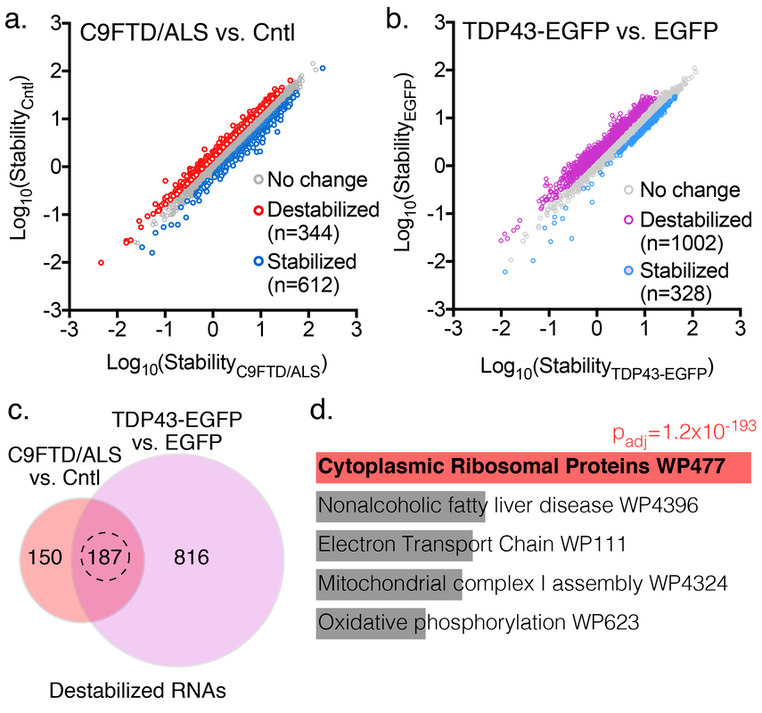
(a) >900 transcripts showed differences in stability in induced pluripotent stem cells (iPSCs) from individuals with familial FTD/ALS vs. controls. (b) TDP43-EGFP overexpression in control iPSCs largely destabilized transcripts. (c) Of 344 destabilized transcripts in C9ALS iPSCs, 187 (54%) were also destabilized by TDP43 overexpression. (d) These transcripts were heavily enriched in ribosomal mRNAs by gene ontology analysis.
|
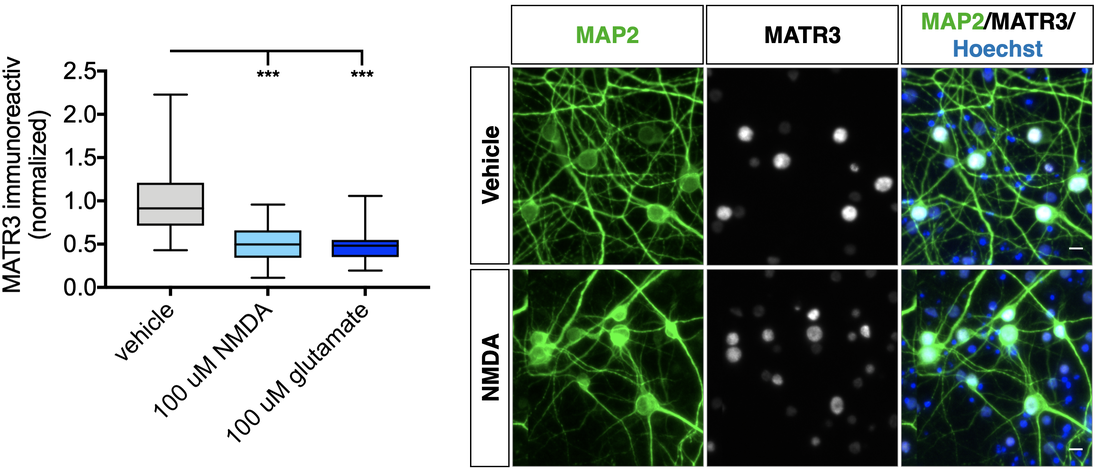
Matrin-3 function and dysfunction
Mutations in the gene encoding the RNA- and DNA-binding protein Matrin-3 (MATR3) underlie familial ALS, dementia, and myopathy, yet we still understand little of the protein's normal function. Our investigations center on mechanisms responsible for regulating MATR3 levels and function in neurons, and how these processes may be disrupted by pathogenic mutations
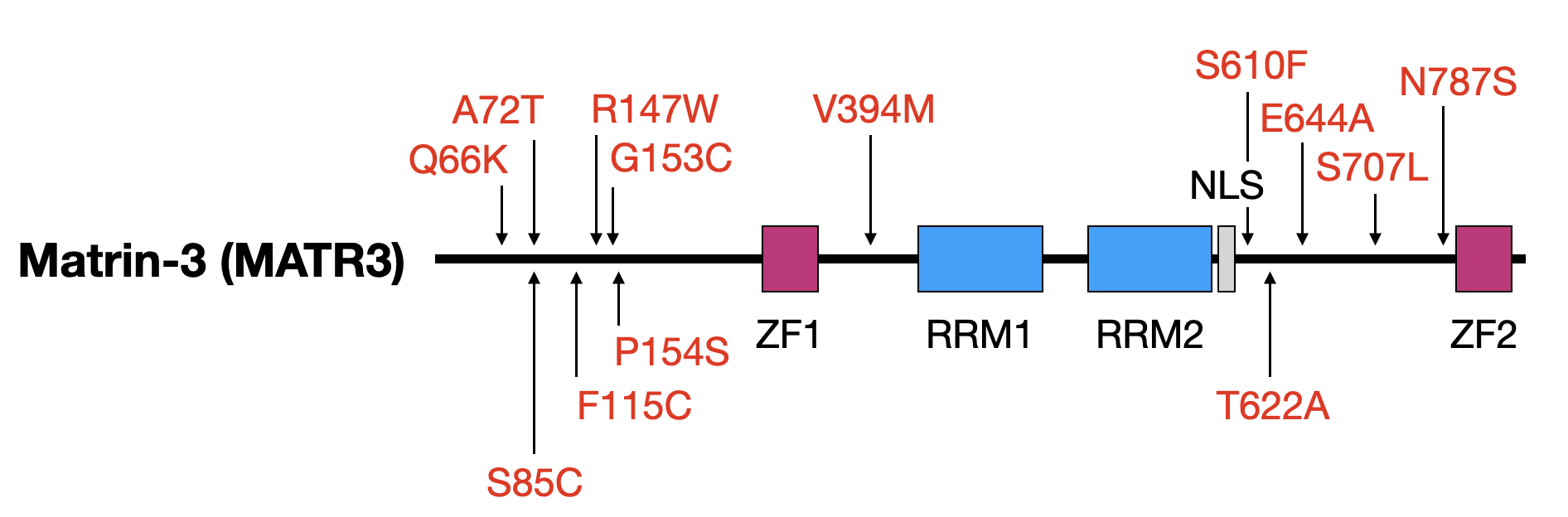
Above — Matrin-3 functional domains and the location of disease-associated mutations. ZF, zinc finger; RRM, RNA recognition motif; NLS, nuclear localization signal.
Below — Quantification of MATR3 immunostaining in MAP2-positive rodent primary cortical neurons after application of NMDA and glutamate for 3 h.
Protein homeostasis in neurodegenerative disorders
The accumulation of misfolded proteins is a universal hallmark of nearly all neurodegenerative disorders, suggesting that in these conditions, neurons are incapable of maintaining protein homeostasis. We are investigating the pathways by which misfolded proteins are degraded specifically in neurons, and also devising novel methods to identify potent activators of neuronal protein clearance mechanisms.
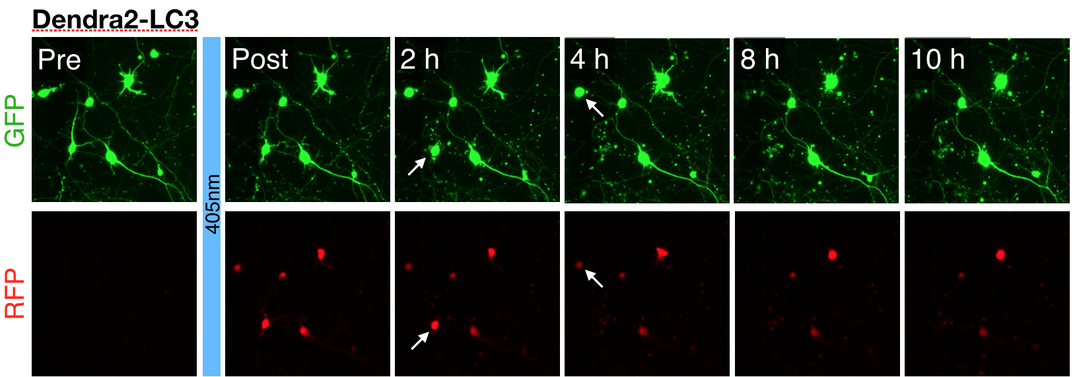
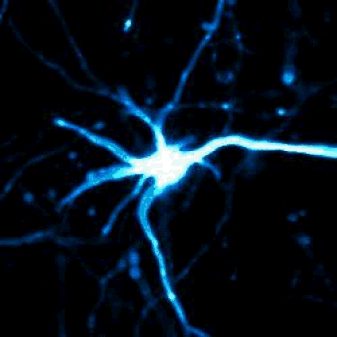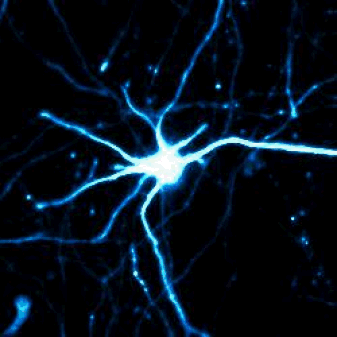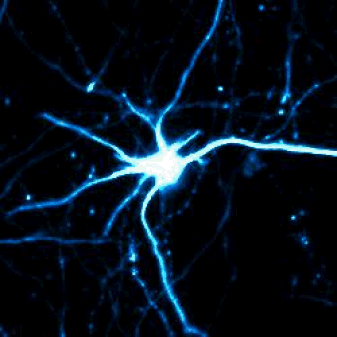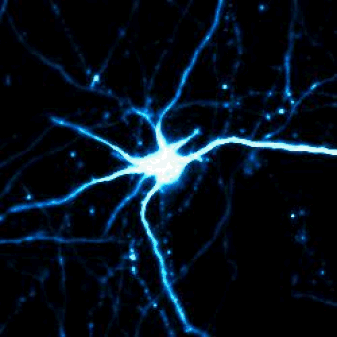
Above — Optical pulse labeling of LC3, a marker and substrate of autophagy. Techniques such as these enable us to track the activity of protein clearance mechanisms in situ.
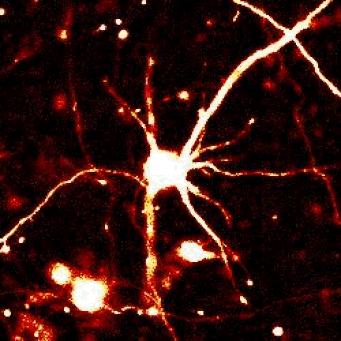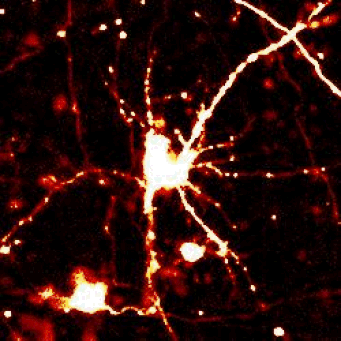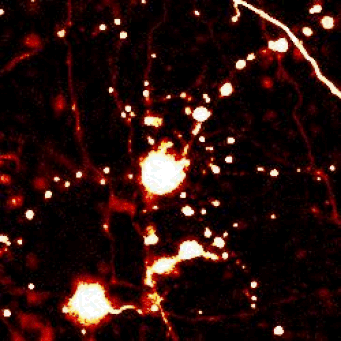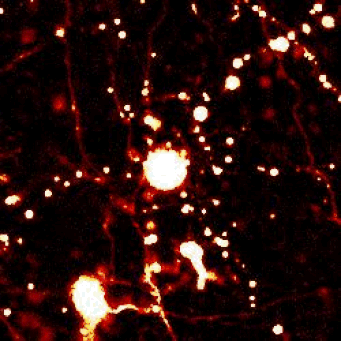
|
Left — A neuron unable to maintain protein homeostasis undergoes apoptosis.
Right — Therapies that activate autophagy or other protein clearance pathways promote neuronal survival in disease models.
|
|
ß-propeller protein associated neurodegeneration (BPAN)
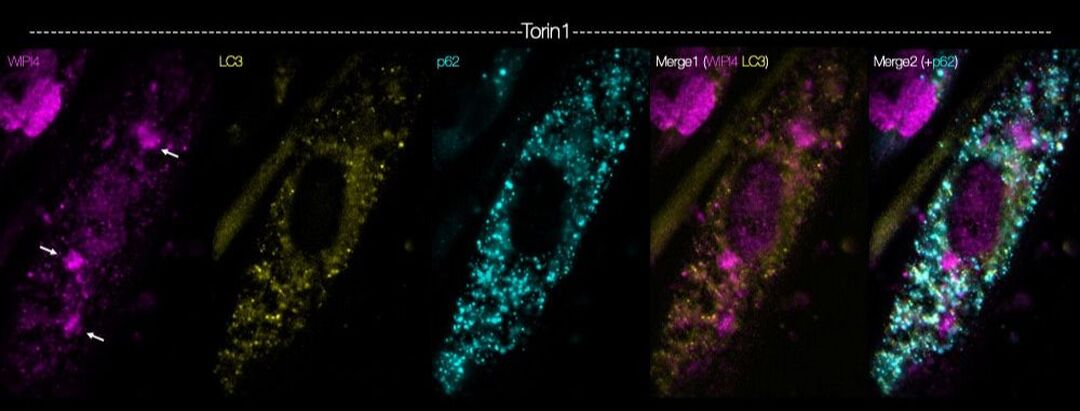
BPAN is an incurable, progressive neurologic disorder, caused by mutations in the X-linked gene, WDR45, which encodes the autophagy-related protein WIPI4. Several critical questions in BPAN remain unanswered; for instance, why do WDR45 mutations cause neurodegeneration despite ubiquitous expression of WIPI4? Do WDR45 mutations lead to neurologic deterioration in BPAN by disrupting autophagy? With support from Don't Forget Morgan, we are addressing these questions by creating novel, induced pluripotent stem cell (iPSC)-based model systems derived from affected patients. Using fluorescent autophagy reporters, CRISPR/Cas9 gene editing, and automated microscopy, we are investigating the neuron-specific functions of WIPI4, how BPAN-related mutations impact neuronal viability, and testing genetic and pharmacologic candidates to identify strategies for preventing BPAN-related toxicity.
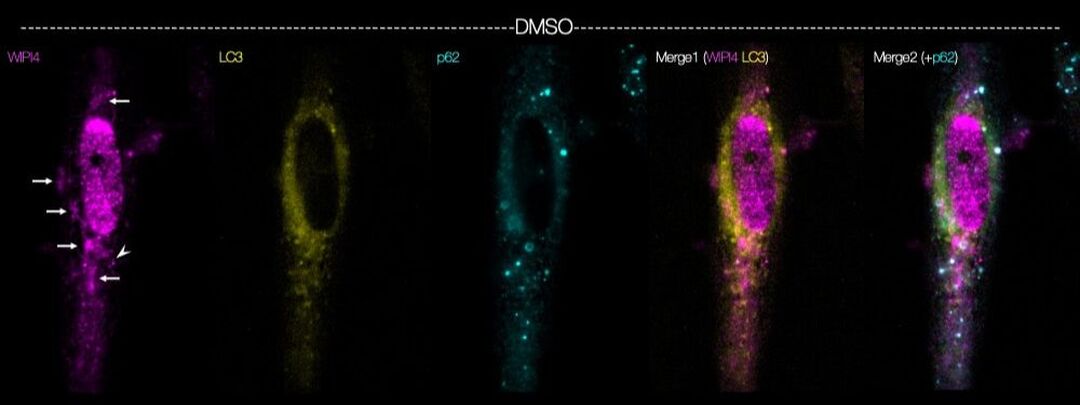
Above — Myocytes differentiated from human induced pluripotent stem cells were immunostained using antibodies against WIPI4 (magenta) and the autophagy related proteins LC3 (yellow) and p62 (cyan).
Below — the same experiment was repeated in the presence of the mTOR inhibitor Torin1.
Disease mechanisms and therapeutic approaches in chorea acanthocytosis
Vacuolar protein sorting 13A (VPS13A) is a large multidomain protein that is highly evolutionarily conserved. VPS13A mutations result in an unusual condition characterized by seizures, parkinsonism, and cognitive decline. Supported through a family-centered foundation, we are investigating the function of VPS13A, the molecular pathways responsible for neurodegeneration in chorea acanthocytosis, and novel neuroprotective strategies.
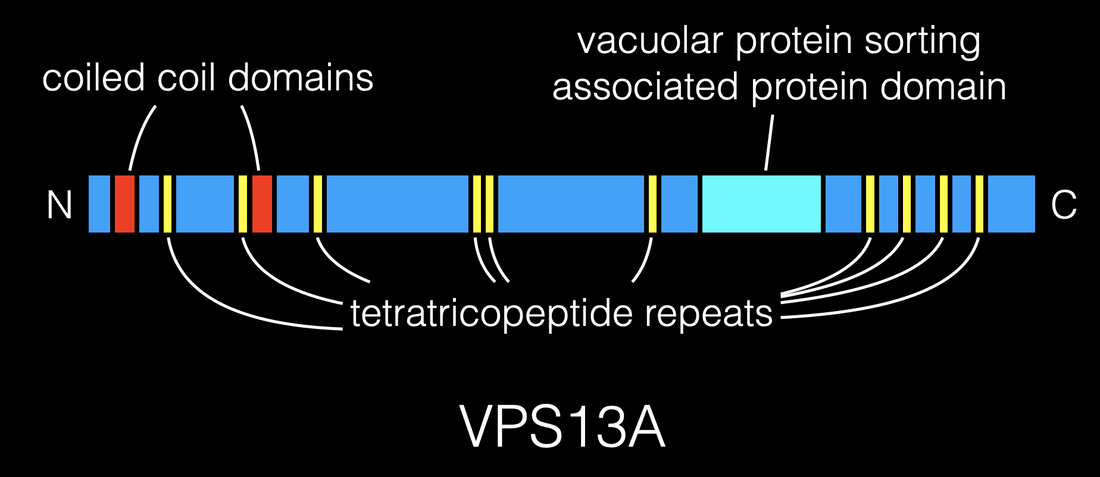
Above — Schematic of VPS13A protein.
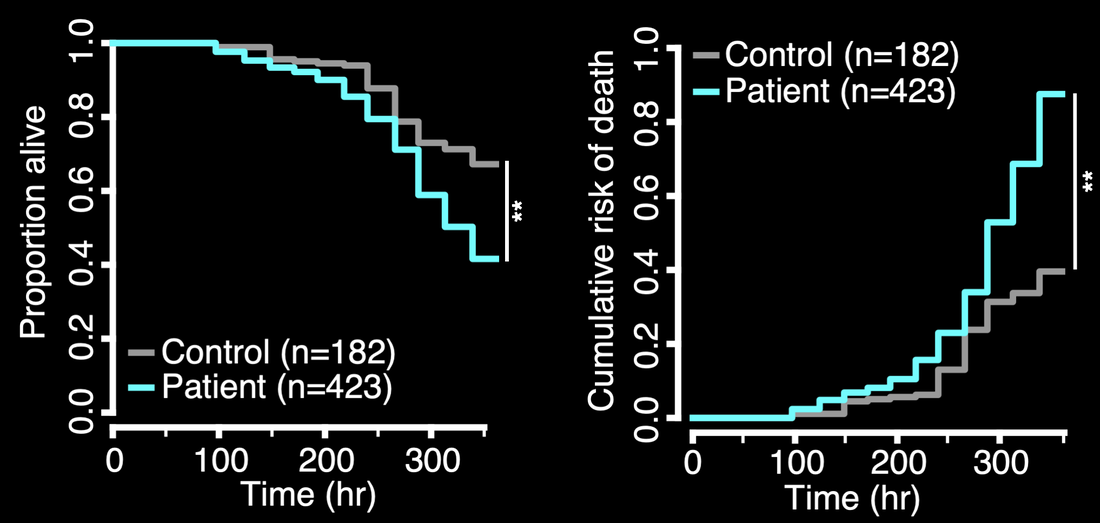
Below — Human iPSCs donated by individuals with chorea acanthocytosis (patient) or unaffected family members (control) were differentiated into neurons and followed by automated microscopy and survival analysis, demonstrating an elevated risk of death in patient-derived neurons.
High-content automated image analysis
What discriminates the neurons that die from those that survive? Can we capitalize on these differences to improve survival and prevent disease progression? We are attempting to answer these questions by developing high-content and high-throughput image analyses algorithms that incorporate structural and functional variables to predict neuronal survival in ALS/FTD models.
|
|
Single-cell shape analysis of primary rodent neurons in culture
|